11 Aqueous Geochemistry
11.2 Overview
The module allows the user to choose from a wide array of geochemical ‘components’ (e.g., anions, cations, metals) to simulate, and then solves for the equilibrium speciation of the solution. The module includes an advanced solver, and provides pH and other solution properties, and optional mineral or gas phases. Relevant kinetic transformations (e.g. redox reactions) between simulated components are also included. Metals may be simulated as part of the model and can also be active within the biological cycles. The module is flexible and simple in its configuration and can be used in the water column and in the sediment if the dynamic sediment diagenesis model (see Section ?) is also being simulated. The module dynamically links with the other biogeochemical processes within AED, such that any biological activity (e.g. algal nutrient uptake and photosynthesis, microbial respiration, and nitrification) will dynamically affect the aqueous speciation and pH. Details of the equilibrium and non-equilibrium processes included are described in the adjacent tab.
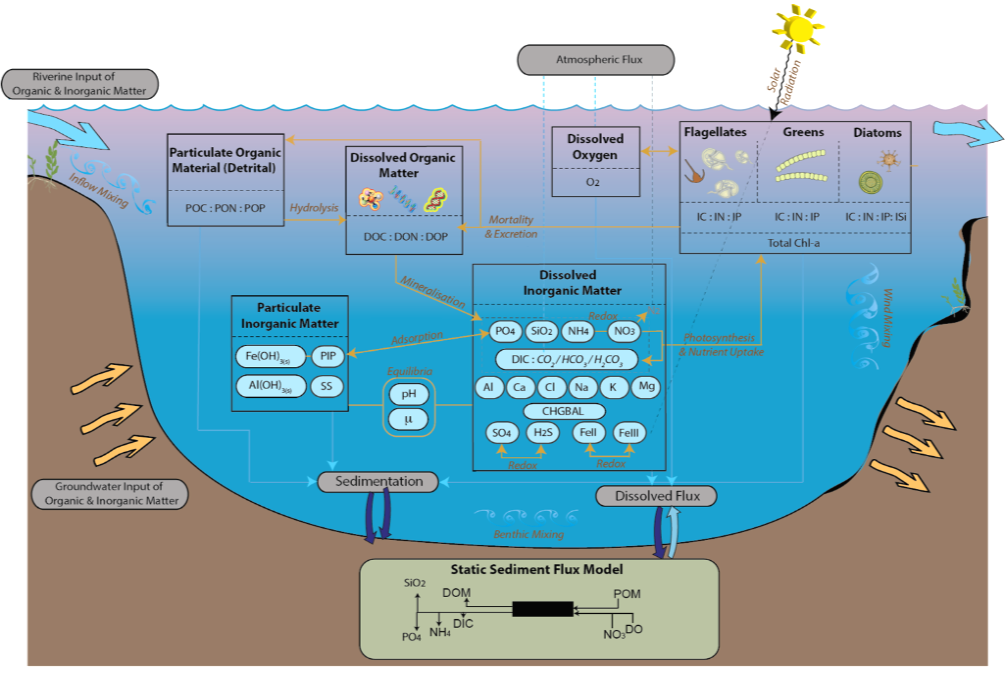
Figure 11.1: Conceptual diagram highlighting key variables and interactions within the model
11.3 Model Description
The aqueous geochemistry sub-model was developed to simulate aqueous speciation and solubility equilibrium control of pure-phase minerals using an approach similar to PHREEQC and as reported previously in Salmon et al (in review). Each dissolved component, \(X\), and mineral phase, \(PP\), is a state variable within the model and is mass-conserved. In addition to external loading from inflows, the model includes a configurable dissolved sediment flux term for the dissolved species and a sedimentation term for the particulates. If we remove the hydrodynamic terms (e.g., inflows/outflows, mixing) for brevity, the differential equation is defined for the dissolved state variables as:
11.3.1 Process Descriptions
Aqueous Speciation & Solubility Equilibrium Control
The aqueous geochemistry model is conceptually similar to other equilbrium codes. The model defines geochemical reactions in terms of components. An aqueous species \(C\) is created as a product of reaction between components \(A\) and \(B\), as shown in the following example:
where \(a\), \(b\) and \(c\) are the stoichiometric coefficients. The reaction shown above is able to proceed back and forward depending on the prevailing conditions of the aqueous solution. The equilibrium of the reaction is a function of the standard free energy, and from this the familiar mass-action expression is defined:
where \(K_{C}\) is denoted the equilibrium constant. In any natural aquatic system, numerous components are present and there are therefore hundreds of reactions similar to Eq. above that will be close to equilibrium. The many reactions form a complex and interdependent set of simultaneous expressions which can be solved numerically, discussed next.
For each component, \(X_{x}\), (generally components are equivalent to elements, except for the case of elements with multiple redox states, in which case each redox state is assigned as a unique component), a set of \(M\) components is defined, \(X \in \{X_1, X_2, ..., X_M\}\). Combination of all the components into an aqueous solution results in numerous reactions taking place of the form of Eq. .., and we define the resultant set of aqueous species that result as: \(C \in \{C_1, C_2 ..., C_N\}\), where \(N\) is the total number produced.
The `activity’ of an aqueous species is not equivalent to its molality due to the nonideality of aqueous solutions; a species activity, is related to its molality according to the activity coefficient, :
Numerous methods exist for estimating a species activity coefficient. The first is known as the Davies equation:
and the other one used in the AED2 solution is the Debye-Huckel equation:
where \(z_{i}\) is the ionic charge of the \(i^{\text{th}}\) species \(c_{1-4}\) are constants that depend upon properties such as the temperature of the solution. The activity of a species is dependent on the ionic strength, \(\mu\), a solution property defined as:
where \(W_{aq}\) is mass of water in the aqueous phase.
To numerically solve the system of equations, the total number of moles of any component is estimated by adding up the amount contained in each species:
where \())aix\) is the stoichiometric coefficient of component \(x\) in species \(i\). The activity of each species is calculated using the mass-action equations, which are defined more generically as:
Taking the logarithm of both sides of Equation above leaves:
which can be defined in matrix notation as:
where \(*\) denotes a vector quantity. The solution is achieved by iterating using a Newton-Rhapson scheme, which aims to minimize the residual, \(R\), between the estimated component molalities (\(T\)) and the known values:
In matrix notation:
The Newton-Rhapson technique iterates towards a solution by employing a derivative (termed Jacobian) matrix, \(J\):
\[\begin{equation} R=J \Delta X \tag{11.13} \end{equation}\]
which is solved for \(\Delta X\) at each iteration. The new estimates for \(X\) are then updated according to:
and the whole scheme proceeds until the solution has sufficiently converged, \(R<\epsilon\), where \(\epsilon\) is a pre-defined convergence criterion (\(10^{-10}\) in this code). The Jacobian matrix is defined as:
which can be expanded to:
using Eq. …. The derivatives are then defined according to:
and
which are the substituted into Eq. .. to leave
Therefore for a given total number of moles of each element in each computational cell, using this scheme the model solves for the activity of each aqueous species, ionic strength and \(pH\). The charge balance variable (denoted \(ubalchg\)) is also subject to advection and mixing as all other state variables, and importantly, an estimate of \(ubalchg\) must be provided at any inflow boundaries. If this is not done, then the charge balance will be compromised, and will manifest in a poor \(pH\) prediction.
The Newton-Rhapson scheme described above is implemented by making use of the Simplex numerical solver (Barrodale and Roberts, 1980; Parkhurst and Appelo, 1999). This is because AED2 also allows for simulation of pure phase (i.e. non-aqueous phase) minerals. When minerals can precipitate and dissolve, properties such as the ionic strength (\(MU\)), activity of water (\(XH_{2}O\)) and charge balance (\(CV\)) may be non-conservative in the aqueous solution. These are therefore included as unknowns in the simplex solver, which uses the Newton-Rhapson technique to solve the following matrix:
The operation of the simplex solver is complicated and the reader is referred to Barrodale and Roberts (1980) for a detailed account. The aqueous species used in the simulations is based on those from Nordstrom et al. (1990), termed the WATEQ4F database.
11.3.3 Parameter Summary
| Parameter Name | Description | Units | Parameter Type | Default | Typical Range | Comment |
|---|---|---|---|---|---|---|
geochem_file
|
Link to geochem database | — | string | — | — | — |
num_components
|
number f dissolved components to be concidered | — | integer | — | — | — |
dis_components
|
list of component names | — | string | — | — | — |
component_link
|
Active variables to be used as a componenet | — | string | — | — | — |
Fsed_gch
|
sediment flux rate of dissolved components | \(mmol\,m^{-2}\,day^{-1}\) | array | — | — | — |
num_minerals
|
number of particulate mineral phases to be included | — | integer | — | — | — |
the_minerals
|
list of mineral names | — | string | — | — | — |
mineral_link
|
name of any active variables to be used as a mineral | — | string | — | — | — |
w_gch
|
sedimentation rate of chosen particulate phases | \(m\,day^{-1}\) | array | — | — | — |
Riron_red
|
— | — | float | — | — | — |
Kiron_red
|
— | — | float | — | — | — |
theta_iron_red
|
— | — | float | — | — | — |
Riron_aox
|
— | — | float | — | — | — |
Riron_box
|
— | — | float | — | — | — |
theta_iron_ox
|
— | — | float | — | — | — |
simEq
|
— | — | boolean | — | — | — |
dis_initial
|
— | — | array | — | — | — |
speciesOutput
|
— | — | string | — | — | — |
min_initial
|
— | — | array | — | — | — |
11.4 Setup & Configuration
11.4.1 Setup Example
An example nml block for the geochemistry module is shown below:
&aed_geochemistry
geochem_file = './AED2/aed_geochem_pars.dat'
!-- dissolved components
num_components = 10
dis_components = 'DIC', 'FeII', 'FeIII' ,'Al', 'Cl', 'SO4', 'Na', 'K', 'Mg', 'Ca','H2S'
component_link = 'CAR_dic','','','','','','','','','','','',''
Fsed_gch = 0.,0.,0.,0.,0.,0.,0.,0.,0.,0.,0.,0.
!-- mineral components
num_minerals = 3
the_minerals = 'Gibbsite','FeOH3A','Calcite'
mineral_link = '', '', ''
w_gch = 0.0, -1.0, 0.0
!-- iron redox
Riron_red = 0.0
Kiron_red = 100.
theta_iron_red = 1.07
Riron_aox = 0.0 !1 = 100% per day decay
Riron_box = 0.5
theta_iron_ox = 1.06
!-- advanced
simEq = .true.
! dis_initial = 0.1,0.1,0.1,0.1,0.1,0.1,0.1,0.1,0.1,0.1,0.1,0.1
! speciesOutput = 'CO2'
! min_initial = 0.01,0.01,0.01,0.01
/aed.nml, users must also supply a valid AED geochemistry reaction database file (aed_geochem_pars). This file must be supplied in the specified DAT format.